Ivanir Ferreira, Jornal da USP
Arte: Moisés Dourado / Jornal da USP
A doença afeta crianças menores de 8 anos (meninas) ou 9 anos (meninos) e está associada a uma mutação da proteína MKRN3

Arte sobre imagens Freepik e Anderson Araújo/CB/DA Press
Uma pesquisa com crianças de várias etnias ajuda a mostrar o pioneirismo de cientistas brasileiros na descrição de mutações do gene MKRN3, localizado no cromossomo 15, atualmente reconhecidas como uma causa frequente da puberdade precoce central (PPC) de origem genética. Nesse trabalho recente, que teve a colaboração de pesquisadores internacionais, foi analisado material genético de 716 crianças de várias regiões: brasileira, americana, argentina, belga, israelense, turca e norueguesa.
No Brasil, as amostras dos pacientes foram estudadas no Laboratório de Hormônios e Genética Molecular (LIM-42) do Hospital das Clínicas (HC) da Faculdade de Medicina da USP (FMUSP), cujos pesquisadores, em 2013, estavam à frente da descoberta da causa da PPC. Os resultados estão no artigo Genotype-phenotype correlations in central precocious puberty caused by MKRN3 mutations, publicado em abril, no periódico The Journal of Clinical Endocrinology & Metabolism.
Os resultados obtidos são importantes porque possibilitam o acompanhamento genético dos pacientes e a identificação de novos casos na família, além de detectar a patogênese das mutações e potencializar o sucesso do tratamento, destaca a médica endocrinologista Ana Claudia Latronico, professora da Faculdade de Medicina da USP e uma das pesquisadoras do LIM-42.
Em geral, a puberdade precoce central caracteriza-se pelo surgimento prematuro de sinais da puberdade. Em meninas, os sintomas incluem o desenvolvimento das mamas, o surgimento de pelos pubianos e nas axilas e a chegada do primeiro período menstrual. Nos meninos, incluem testículos e pênis aumentados, voz grossa e pelos faciais, pubianos e axilares.
“Há também um avanço da maturação óssea (fechamento precoce dos espaços ósseos), o que resulta em uma baixa estatura na vida adulta se não houver tratamento adequado, em torno de 10 a 20 centímetros abaixo do estimado, segundo o perfil da família”, explica ao Jornal da USP a endocrinologista Ana Claudia Latronico.
Até 2013, não se sabia exatamente a causa mais frequente da forma familiar de PPC. A descrição das mutações do gene MKRN3 realizada pelos pesquisadores brasileiros levou ao diagnóstico mais assertivo e tratamento mais precoce.
Além de impactar aspectos físicos, existem repercussões emocionais nas crianças afetadas pela doença. “Há um sofrimento psíquico pela percepção das diferenças físicas em relação aos pares do mesmo sexo e idade”, relata a pesquisadora.
A médica Ana Claudia tem larga experiência na área. Além de orientar inúmeras pesquisas sobre o assunto no LIM-42, há mais de 25 anos atende pacientes com características clínicas sugestivas de antecipação do desenvolvimento da puberdade no Ambulatório do HC.
O diagnóstico é baseado nas evidências clínicas, exames laboratoriais e de imagens: ultrassom pélvico nas meninas, para avaliação do desenvolvimento dos ovários e do útero; e ressonância magnética de crânio para avaliar risco de lesões tumorais em ambos os sexos.
O tratamento é realizado com medicamentos que bloqueiam a evolução da puberdade, promovem a regressão dos caracteres sexuais secundários e diminuem a velocidade do crescimento e a progressão da idade óssea.
Até 2013, não se sabia exatamente a causa mais frequente da forma familiar de PPC. A descrição das mutações do gene MKRN3 realizada pelos pesquisadores brasileiros levou ao diagnóstico mais assertivo e tratamento mais precoce.
Pulsos antecipados dos hormônios da puberdade
Sobre o funcionamento do MKRN3, a endocrinologista explica: “Ele é produzido na região do hipotálamo e tem ação supressora (inibe) na produção do hormônio GnRH (hormônio liberador das gonodotrofinas), que tem um papel-chave no eixo hormonal reprodutivo humano”. Durante a infância, o GnRH é silenciado e só deveria começar a ser secretado no início da puberdade, por volta dos 10 ou 11 anos de idade.
Segundo a pesquisadora, a partir desse período, o hormônio GnRH é liberado de forma pulsátil, com frequência e amplitude aumentada, para que a hipófise seja estimulada a produzir gonodotrofinas (hormônios luteinizante – LH e folículo-estimulante – FSH) que, por sua vez, estimulam a produção de estrógeno no sexo feminino e a testosterona no sexo masculino, resultando no surgimento dos caracteres sexuais secundários, diz.
Se o MKRN3 estiver mutado, ele perde a capacidade de inibir o GnRH, que começa a ser secretado no organismo do indivíduo ainda na infância. “A ativação precoce do eixo hipotálamo-hipófise-gonadal devido à liberação do GnRH resulta em puberdade precoce central”, diz.
A mutação genética do MKRN3 é herdada sempre pelo lado paterno. Quando a mutação está do lado materno, o alelo é silenciado. A mãe pode ser uma carreadora assintomática. Ela pode eventualmente passar para um filho e esse filho ter filhas ou filhos com puberdade precoce. Uma mulher carreadora pode, portanto, ter netos com a patologia.
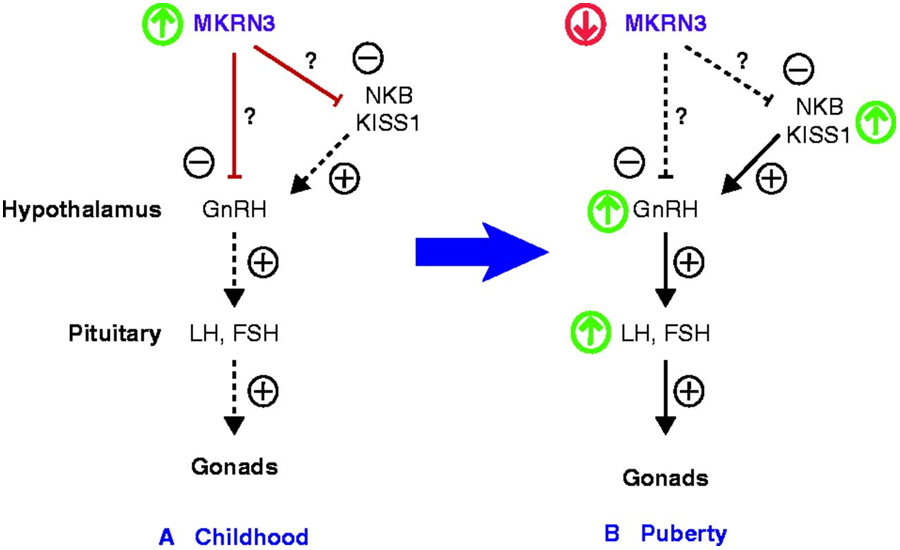
O MKRN3 mutato perde a capacidade de inibir o GnRH, que começa a ser secretado no organismo ainda na infância, levando ao surgimento precoce de sinais puberais – Fonte: Abreu AP et al., Lancet 2016
Pesquisa ampliada: grande coorte e pacientes multiétnicos
Até 2013, a causa da puberdade precoce central era desconhecida. “Hoje, a doença tem causas genéticas bem estabelecidas e prevalentes, graças à descrição, realizada por brasileiros, de diferentes tipos de mutações [frameshift, stop gain e missense] que afetam a proteína MKRN3 ”, relata ao Jornal da USP a professora Ana Claudia.
Em um artigo anterior, Central Precocious Puberty Caused by Mutations in the Imprinted Gene MKRN3, publicado na revista The New England Journal of Medicine, em junho de 2013, os cientistas analisaram o sequenciamento total do exoma (fração do genoma) em 40 membros de 15 famílias com puberdade precoce central. Depois disso, outros trabalhos brasileiros e estrangeiros, incluindo uma metanálise (avaliação de várias pesquisas independentes sobre a mesma questão), confirmaram o pioneirismo dos pesquisadores do LIM-42.
O artigo mais recente, Genotype-phenotype correlations in central precocious puberty caused by MKRN3 mutations, com os resultados de estudo de casos multiétnicos e com uma grande coorte de pacientes, mostrou que as mutações de perda de função de MKRN3 representam a causa genética mais prevalente de PPC (de 33% a 46%).
Ao todo, foram analisadas amostras de DNA e dados clínicos de 716 crianças com PCC; sendo que 517 foram estudados por pesquisadores do LIM-42. Desse montante, em 71 pacientes foram detectadas mutações inativadoras do MKRN3 – 40 eram brasileiros, nove americanos, oito espanhóis, cinco argentinos, quatro belgas, dois israelenses, um turco e um norueguês.
Se o MKRN3 estiver mutado, ele perde a capacidade de inibir o GnRH que começa a ser secretado no organismo do indivíduo ainda na infância. “A ativação precoce do eixo hipotálamo-hipófise-gonadal devido à liberação do GnRH resulta em puberdade precoce central”, diz.
Dos 71 pacientes com mutações inativadoras do MKRN3, 45 foram encontradas em amostras de DNA de meninas e 26 de meninos, oriundos de 36 famílias (havia mais de um caso por família). Destes pacientes, 18 tiveram mutações diferentes de perda da função MKRN3; e oito foram classificados como graves (mutações do tipo frameshift e stop códon foram presentes em 70% dos pacientes).
Das crianças investigadas, os primeiros sinais puberais ocorreram aos 6,2 anos nas meninas e 7,1 anos, nos meninos. Os 53 pacientes que abrigavam mutações graves no MKRN3 tiveram maior avanço da idade óssea do que aqueles com mutações do tipo missense (que gera a troca de aminoácido na proteína). E também apresentaram níveis mais elevados de hormônio LH basal (responsável pelo amadurecimento dos folículos, ovulação e produção de progesterona nas meninas).
Pacientes com PPC causada por mutações no MKRN3 tinham FSH mais elevado e menor intervalo entre o início dos sintomas e a busca a um serviço médico (primeira consulta). Em meninos, o diagnóstico se deu através de rastreio familiar em 87,5% dos casos e não consulta familiar.
O trabalho de coorte multiétnica envolvendo 716 amostras genéticas de crianças de vários países teve apoio financeiro da Fundação de Amparo à Pesquisa do Estado de São Paulo (Fapesp) e do Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
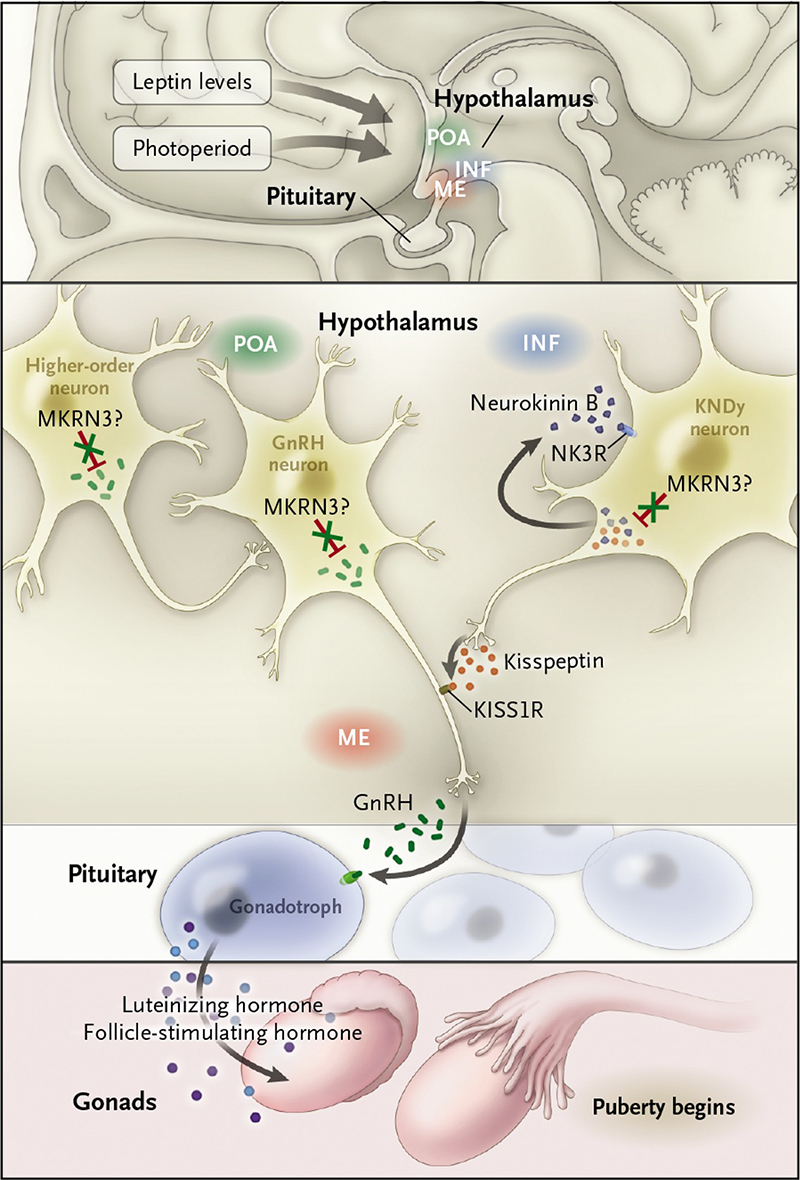
Um evento fundamental no início da puberdade em mamíferos é a retomada da liberação pulsátil do hormônio liberador de gonadotrofina (GnRH) dos neurônios do hipotálamo – Fonte: Abreu AP et al. NEJM 2013